会员投稿
会员投稿 手机版
手机版
黄选洋 译 张配配 校
摘 要:全基因组序列数据的使用在家畜育种计划中具有巨大的潜力,可以提高发现变异基因的能力,同时能更准确和更持久地预测育种值而不是标记阵列。要了解家畜基因组序列数据的全部潜力,需要从大量的个体,甚至要从数百万个个体上获得基因组序列和表型数据,从而准确地估测构成数量性状基础的大量致病变异的影响。
关键词:全基因组;标记阵列;估测;变异;杂交剥离
低成本的测序策略结合估测法(imputation),能够以负担得起的成本为大量个体生成所需基因组序列的信息。低覆盖率使研究人员对大量个体进行基因组测序成为可能,这可以提高变异的发现率,特别是低频率的变异,并能加强根据基因组序列数据对整个群体的估测。
本文介绍了我们在一项研究中所采用的策略,该研究对来自9个商业品系的7 848头猪进行了全基因组测序,这些品系大部分处于低覆盖率范围。随后,我们证明,将该测序策略与“杂交剥离”估测法相结合,是一种可为大群家畜纯种系谱产生全基因组序列数据的有效策略。最后,我们测试了这些大数据集对合成表型的基因组预测的优势。
1 材料和方法
1.1 测序策略
我们对Genus plc公司的9个商业品系(PIC猪商业品系,公司位于美国田纳西州亨德森县)的7 848头猪的全基因组进行了测序。测序时,我们从每个品系中选择约2%(1.7%~2.5%)的猪。结果表明,大多数猪处于低覆盖率,目标覆盖率为1倍或2倍,一小部分猪处于较高的覆盖率,分别为5倍、 15倍或30倍。个体的平均覆盖率为4.1倍,但中位数为1.5倍。我们使用三步策略选择个体和这些个体的覆盖范围:
第一步:在纯种系谱中贡献最多基因型后代的父系和母系分别拥有2倍和1倍的覆盖率。
第二步:AlphaSeqOpt法第1部分用于识别在种群单倍型中占有最大比例的单倍型个体,并在控制总成本的前提下,为它们及其祖先分配一个介于0倍至30倍的最优水平的测序覆盖。
第三步:AlphaSeqOpt法第2部分用于识别累计覆盖率低(低于10倍)的单倍型个体,并对这些个体进行1倍测序,以增加单倍型的累计覆盖率(即大于或等于10倍)。
AlphaSeqOpt法使用根据阶段性标记阵列基因型推断的单倍型。
1.2 发现变异
将测序结果与Sscrofa 11.1参考基因组进行比对,利用一个基于GATK 3.8的Haplotype-Caller工具的数据来源找出变异。为了避免在应用低覆盖率序列数据时对GATK引入的参考等位基因产生误差,我们利用堆积函数提取了支持该等位基因的读取数,结果从这9个品系中共发现了6 000万个单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)。
1.3 估测全基因组序列数据
使用商业标记阵列对每个群体中的大多数个体进行基因分型,拥有15 000个低密度(Low Density,LD)或75 000个高密度(High Density,HD)全基因组标记。正如用AlphaPeel法测算的那样,采用杂交剥离估测法分别估测每个群体的全基因组序列。该方法通过两阶段,降低估测成本:
• 多轨迹迭代剥离,可以根据数组中的该标记估计分离概率。
• 改进的单位点迭代剥离,可以基于序列数据旁侧数组的该标记的估测值,利用该序列数据大致估计任何其他变异位点上的分离概率。由于每条染色体中重组基因的数量有限,以及附近标记共同被遗传的概率很高,这种大致估测的精度损失可以忽略不计。9个品系估测出的猪总数约为35万头。
为了评估估测的准确性,我们使用了来自4个大小不同的群体在高覆盖率(15倍或30倍)下测序的284个个体。被检测个体的序列数据用留一法设计(leave-one-out design)可以完全掩盖。将估测的等位基因剂量与获得完整数据的等位基因剂量进行比较,认为是“真”值。
1.4 基因组的预测
我们在一个拥有3万个个体的品系中检测了基因组预测的准确性,这些个体的估测基因型为1 600万个SNPs。正如在AlphaBayes软件中预测的那样,使用岭回归( ridge regression)模型预测基因组。
利用该模式测试了22 318个个体,验证了1 458个个体。对9个具有不同遗传力和数量性状核苷酸(Quantitative Trait Nucleotides,QTN)的合成性状进行基因组预测。
使用4组标记进行基因组预测:从阵列中预选5.7万个标记(HD),从基于LD修剪的序列数据中预选24.8万个变体[全基因组测序(Whole Genome Sequencing,WGS)_LD,WGS_LD],从基于单标记回归结果[(WGS_基于总数据的孟德尔随机化(Summary data-based Mendelian Randomization,SMR),WGS_SMR]的序列数据中预选18.3万个变体,或通过仅每保留第200个变体(WGS_200)从该序列数据中预选6.7万个变体。基因组估计育种值(Genomic Estimated Breeding Value,gEBV)的准确性是根据该验证数据集中gEBV与合成表型之间的相关性来估计的。
2 结果和讨论
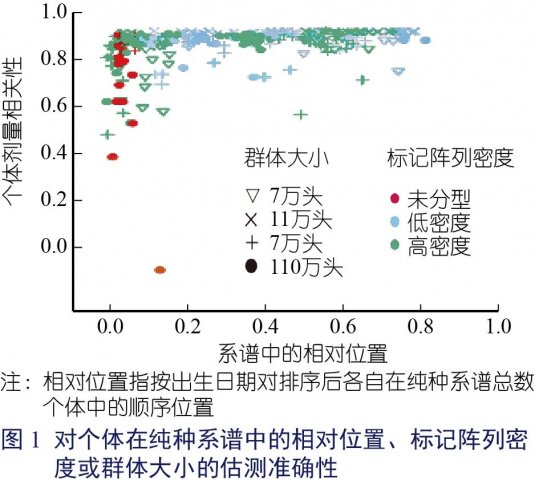
2.1 估测的准确性
对大多数受试个体而言,真实数据的估测精度较高(图1)。平均个体剂量相关性为0.94,中位数为0.97,四分位数范围为0.94~0.98。一些属于该纯种系谱最早几个世代的最古老的个体(位于系谱的前20%)具有很低的估测精度,因为它们无法提供其直系祖先的信息,或能够提供的信息极少,这影响了估测精度。

较晚几个世代的个体(位于系谱中前20%的后面)有更高的估测精度,平均剂量相关性为0.97,变异性更低:中位数为0.98,四分位数间距为0.96~0.99。
个体的标记阵列密度与用标记阵列基因分型获得的直系祖先的数量相矛盾,但对稍后几个世代的个体而言,标记阵列密度的HD和LD之间无显著差异,种群大小对估测精度的影响无明显的倾向性。
2.2 基因预测
在某些情况下,与标记阵列相比,序列数据能够提供更好的预测精度,但其优势取决于该性状的遗传结构。
表1列出了9个合成性状的基因组预测精度。当QTN的数量较小时,可以识别能支撑该性状的遗传变异的变体(variants)具有足够的统计功效(statistical power),使用这些变体(WGS_SMR)进行预测的准确性高于用来自商业标记阵列(HD)的标记进行预测的。这与之前的观察结果一致,添加一个或几个具有较大作用的标记作为预测因子可以提高该标记序列的预测精度。

当QTN的数量较大时,WGS_SMR的性能比HD的差。在这种情况下,从序列数据中选择的其他变异集可能(略微)比商业标记序列更有利,因为它们不会像商业标记序列那样受到确定偏倚(ascertainment bias)的影响。
这些结果部分是由于目前使用商业标记阵列进行基因组选择已经获得了很高的预测准确性,且与其他研究结果一致。后者发现,与HD标记阵列相比,序列数据在基因组预测上没有改善或只有微小的变化。有待确定的是,结果是否会因以下原因而得到改善:来自多个品种的数据,使用多品种测试和更大的测试集,或比岭回归更适合于大规模开发序列数据的基因组预测方法。
3 结论
无论种群的规模多大,只要个体与具有标记阵列或序列数据的亲缘联系在一起,同时该亲缘有足够多的信息,恰当的测序策略和“杂交剥离”的结合是在大群的纯种系谱中生成全基因组序列数据的一种有效方法。
目前尚不清楚,这些带有估测序列数据的大数据集是否能够提高基因组预测的准确性。

感谢您的支持,我会继续努力的!



打开支付宝扫一扫,即可进行扫码打赏哦